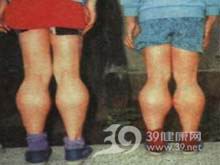
良性假肥大型肌营养不良症详述
(一)发病原因
本症为遗传性疾病,多属性连锁隐性遗传,个别为染色隐性遗传。
(二)发病机制
应用分子生物学方法已将DMD的基因定位于X染色体Xp21.1~Xp21.3,致病基因为dystrophin基因,它是至今发现的最大的人类基因,约2000kb以上,含有79个外显子编码,1个14kb的转录区。研究表明60%~70%的DMD是由于基因缺失或重复突变所致。
基因缺失呈非随机性分布,主要发生在基因的中央区(80%),少数发生在5端(20%)。大的基因缺失常常开始于基因的5端,基因缺失造成开放的读码框的破坏,导致DMD表现。BMD患者,缺失基因保持了翻译读码框,并能产生一个具有一半功能、长度缩短的蛋白质。“读码框”假说解释了92%的DMD/BMD患者不同的临床类型。
dystrophin是dystrophin糖蛋白复合物(DGC)的一部分,DGC是膜相关蛋白的综合体,跨越肌纤维膜,连接细胞内的细胞骨架和细胞外的基质。duchenne肌营养不良患者,由于dystrophin的缺失导致DGC成分的减少,虽能正常合成但不能正确的装配和整合至肌纤维膜上。由此推测由于DGC的受损,引发一系列连锁反应,导致DMD的肌细胞坏死。dystrophin的缺乏使肌纤维膜下的细胞骨架和细胞外基质的联系受到破坏,造成肌纤维膜不稳定,膜撕裂,肌细胞坏死。
良性假肥大型肌营养不良症就诊指南
良性假肥大型肌营养不良症就诊指南针对良性假肥大型肌营养不良症患者去医院就诊时常出现的疑问进行解答,例如:良性假肥大型肌营养不良症挂什么科室的号?良性假肥大型肌营养不良症可能患上什么疾病?与良性假肥大型肌营养不良症容易混淆的症状?医生一般会问什么?等等。良性假肥大型肌营养不良症就诊指南旨在方便良性假肥大型肌营养不良症患者就医,解决良性假肥大型肌营养不良症患者就诊时的疑惑问题。
- 建议就诊科室
- 内分泌科、神经内科、营养科
- 可能疾病
- 1、 肌营养不良症,可能伴随不能露齿及突唇、肌肉假肥大、肌肉肥大等症状,应去神经内科或脑外科就诊。
- 2、 糖原贮积病,可能伴随肌肉萎缩、肝脏肿大、肝肿大等症状,应去内分泌科或儿科就诊。
- 3、 神经系统遗传病,可能伴随不自主运动、发音障碍、弓形足等症状,应去神经内科就诊。
- 4、 进行性肌营养不良症,可能伴随表情淡漠、易跌倒、声音变低等症状,应去神经内科就诊。
- 相关检查
- 1、身体器官功能与营养,身体器官功能与营养是与食物的摄入、消化、吸收和代谢等因素密切相关,好坏可作为鉴定和疾病程度的标准之一,也是诊断身体器官功能的标准之一。
- 2、周围血中异型细胞,周围血中异型细胞检验是神经系统细胞学检查的一种,常用于类脂质沉积病、Ⅱ型糖原沉积痛、β-脂蛋白缺乏病、进行性肌营养不良症进行诊断和鉴别诊断。
- 3、肌电图,通过肌电图检查可以确定周围神经、神经元、神经肌肉接头及肌肉本身的功能状态。
- 4、浆膜腔积液一般性状,出现积液本身就是疾病的一种临床表现。
- 5、血清载脂蛋白AⅡ,测量载脂蛋白AⅡ的含量,降低可见于冠心病、糖尿病、肾病综合征、遗传性ApoA-Ⅰ缺乏症(Tangier病)、家族性低α-脂蛋白血症、鱼眼病、重度营养不良、肝炎活动期、肝功能低下。
良性假肥大型肌营养不良症常见检查
4353 人阅读过 复制文章链接